 A Practical Guide to Protein Crystallization
A Practical Guide to Protein Crystallization A Practical Guide to Protein Crystallization
by Mark Knapp and Bernhard Rupp - in revision
A) Crystallization
It is the goal of the crystal grower to produce resonably large ( >0.1 mm in its smallest dimension) protein crystals, in which the molecules have sufficient long range order that an x-ray beam will be diffracted into a pattern of reflections that will that can ( through crystallographic data analysis) produce a three dimensional map of the molecule's electron density. A model of the protein's known sequence is then fit to this density map. Thus the model structure is dependent completely on the data produced in the xray experiment and that in turn can only be as good as the crystals from which it taken.
In order to obtain a crystal, the protein molecules must assemble into a periodic
lattice. One starts with a solution of the protein with a fairly high concentration
(1.5-200 mg/ml) and adds reagents that reduce the solubility close to spontaneous
precipitation. By slow further concentration, and under conditions suitable for the
formation of a few nucleation sites, small crystals may start to grow. Often very many
conditions have to be tried to succeed.
1) Solubility curve
2) From saturation to crystals
3) Approaching the point of precipitation/crystallization slowly
4) Molecules fitting together like a 3D puzzle (image of molecules in a unit cell).
B) Techniques and tools used in crystallization experiments

Vapor Diffusion
By far the most popoular technique is that of vapor diffusion. In this technique a drop containing protein, stabilizing buffers, precipants and crystallization aids is allowed to equilabrate in a closed system with a much larger reservior. The reservior usually contains the same chemicals minus the protein but at an over all hihger concentration so that water preferencially evaporates from the drop. If conditions are right this will produce a gradual increase in protein and pptnt concentrations so that a few crystals may form.
The conditions which will bring about crystallization are quite varied. One must depend to a great extent on the prior experience of oneself and others. Hampton Research has developed a standard set of screening reagents help enormormously in the initial trials. We have implemented a web computing service version of Brent Segelke's CRYSTOOL, an inherently efficient random screen for crystallization conditions which is constomizable.
Vapor diffusion is performed in one of two general set ups. The one most often used is
called Hanging Drop  in this
diagram one can see that the above mentioned drop is placed on a glass coverslip. This is
then inverted and used to seal a small reservior in Linbro Plate.
After a period of several hours to weeks microscopic crystals may form and may continue to
grow. The other set up is known as Sitting Drop. In this method a drop
usually >10uL is placed in a depression in either a Micro
Bridge in a Linbro Plate or a glass plate and again put into a closed system to
equilibrate against a much larger reservior. The effetct is the same. One usually chooses
a sitting drop if the drop has very low surface tension, making it hard to turn upside
down or if the drops need to be larger than 20uL. Also in some cases crystals will just
grow better in one rather than the other.
in this
diagram one can see that the above mentioned drop is placed on a glass coverslip. This is
then inverted and used to seal a small reservior in Linbro Plate.
After a period of several hours to weeks microscopic crystals may form and may continue to
grow. The other set up is known as Sitting Drop. In this method a drop
usually >10uL is placed in a depression in either a Micro
Bridge in a Linbro Plate or a glass plate and again put into a closed system to
equilibrate against a much larger reservior. The effetct is the same. One usually chooses
a sitting drop if the drop has very low surface tension, making it hard to turn upside
down or if the drops need to be larger than 20uL. Also in some cases crystals will just
grow better in one rather than the other.
Dialysis Techniques
Another method used to bring about gradual change in the relative concentrations of the protein and pptnt is dialysis. This ranges from traditional dialysis tubing for milliliter samples to the use of specialized dialysis buttons which make it easy to dialyse as little as 10uL of sample (illustration). The use of dialysis permits one to increase pptnt level while essentially holding protein and other component concentratons steady. It is also possible to produce crystallization by change in pH by dialysing with a pH gradient.
C) The need for a very pure
protein
The potential to grow exceptionally good, well ordered protein crystals is there only if very pure protein is available. Protein
< 95% pure is not likely to produce very good crystals, if it crystallizes at all. A
general list of contaminants follows:
Contaminants are: unfolded protein, peptides, particulate matter, anything that causes destabilization of the protein, any chemical that is unnecessary for protein stability/solubility or is present in an unnecessarily high concentration.
The protein solution should contain only those chemicals necessary to maintain protein stability and at the lowest concentrations possible. If the protein is stable in Milli Q water, thats the way it should be worked with. In this way one can have the best control and knowledge of the crystallization conditions.
When protein molecules come together to form a crystal it is as if you are trying to build a regular structure with similar but irregularly shaped pieces of jello. The neat little concavities or sites of localized charge can easliy be obscured contaminants. If the protein is contaminated by impurities the molecules will not be able to pack into the crystal lattice as nicely as without.
IF IT SHOWS UP ON A GEL AND ISN'T YOUR PROTEIN, THEN IT'S A CONTAMINANT.
Corollary:
IF A CONTAMINANT SHOWS ON A GEL, THEN THE PROTEIN IS TOO CONTAMINATED FOR
CRYSTALLIZATION.
( But if it's all you have, I'll try it. )
D) Tags used in purification
In these modern times it is now possible to put Tags at either or both the C or N terminus to make purifaiction a much simpler and less toilsome task. This is good because it can greatly improve the end purity of the protein. But one must bear in mind that these tags are probably disordered, just flailing about, rather than joining in the tertiary structure of the molecule. Some there is potential for a
1) His tags - As noted in the link there have been some successes in crystallizing His -Tagged proteins, enough so that the current consensus to try it first with the tag on. Hampton Research has conducted a poll clicke here for the comments.
2) S-tags - there are no literature references to crystallization with these tags. However, a 21 amino acid tail at the end of a protein is a significant addition the terminus and I think bound to interfere with crystallization. ( But I am willing to try it.)
3) Crystallization is easier with a compact molecule that does not have loose ends.
E) The need for a well defined protein.
(This means knowing a lot about it and communicating that to the crystal grower.)
One of the worst things that can happen is for a crystallographer to be handed an ultra
pure sample of concentrated protein and then to be left to find out on his or her own how
easily the protein denatures, or aggregates, or sticks to membranes, dialysis tubing or
any other of the multitude of ways there are to lose protein. Communicate with your
crystallographer.
The following need to be known:
Physical Properties
a) Does it aggregate?
b) Does it adhere to columns or membranes?
c) How long is it stable after isolation?
Chemical Properties
a) known internal disulfide linkages
b) known surface cystienes
c) are metal ions a part of the active protein
What is known about the protein's stability and solubility
a) What's necessary to keep it stable
b) Temperature
c) Additives (Metal cations, EDTA, DTT, BME , K vs. Na .....)
d) Known destabilizing chemicals and conditions
Sequence and common name
a) a list of references dealing with the isolation, activity and purification of the protein
F) Detergents, Buffers and other additives
It is more often than not necessary to prepare a protein solution which contains buffers
and a specific salt content to maintain the proper pH and ionic strength for a protein's
stability. Detergents, particularly in the cases of membrane proteins and DNA repair
enzymes, are also sometimes needed to keep a protein soluble and free from aggregation.
This can be especially true during the crystallization process as the protein
concentration will locally reach extremely high concentrations.
So a few general rules should be followed.
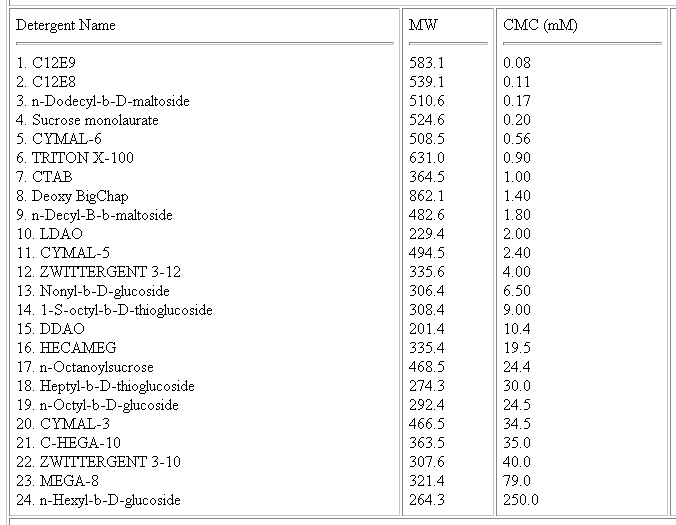
Detergents
a) Keep at or below the CMC
b) As low a molecular weight as possible
c) a nice list of detergents commonly used in
crystallization experiments.
Buffers
a) Keep the concentration as low as possible.
b) Most buffers are fine.
Additives and Salts
a) Glycerol - tends to make the protein too soluble for easy crystallization.
b) Other compounds used for solubilization - Avoid using these if at all possible.
c) Keep salt levels to minimum. These may oversolublize the protein.
Summary
Given high purity protein, at high concentrations, and
enough information, one may be able to grow protein crystals which will lead to a
successful and accurate solution to the protein's structure. Then you can have pretty pictures like these. And nice structures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}