 An
introduction to X-ray crystallographyAn
introduction to X-ray crystallography
An
introduction to X-ray crystallographyAn
introduction to X-ray crystallography
Outline of the experiment
In a macromolecular X-ray diffraction experiment a small protein crystal is placed into an intense X-ray beam and the diffracted X-rays are collected with an area detector (it is advantageous to cool the crystals to low temperatures, primarily to prevent radiation damage). The diffraction pattern consists of reflections of different intensity, and a lot of patterns need to be collected to cover all necessary crystal orientations. After some data processing, we end up with a list of indexed reflections and their intensities.
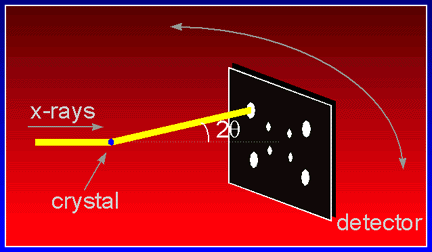
The diffracted X-rays are scattered by the crystal at a certain angle. The further backwards the x-rays scatter, the higher we say is the resolution of the data set. The extent to which the crystal diffracts determines how fine a detail we can actually distiguish in our final model of the structure. High resolution is thus desirable. Knowing the wavelength and the diffraction angle of a reflection, its resolution d can be easily calculated :
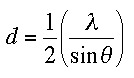
This is just a reformulation of the famous Bragg
equation ![]() .
.
 Click on the phenylalanine to learn more about resolution
Click on the phenylalanine to learn more about resolution
![]() To
learn more about protein crystals, click on the little image
To
learn more about protein crystals, click on the little image 
![]()
![]() Back to X-ray Facility Introduction
Back to X-ray Facility Introduction
LLNL Disclaimer
This World Wide Web site conceived and maintained by
Bernhard Rupp (br@llnl.gov)
Last revised November 02, 1999 16:59
UCRL-MI-125269