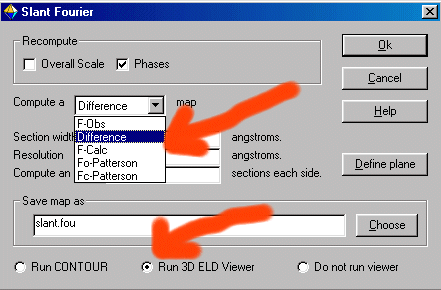
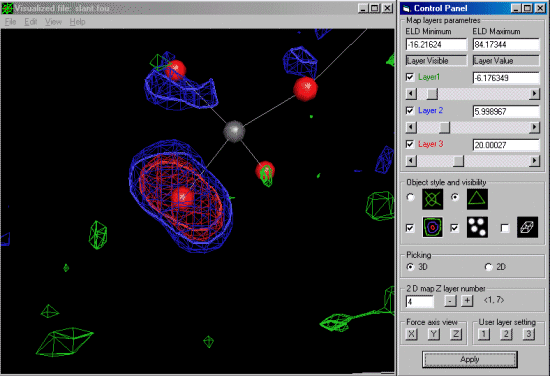
The trick here to get the difference map on the offending atom with the large
thermal is to give it a zero occupancy. Thus it will show up as the primary atom in the
difference map.
The script method of doing this would be to use the following
\edit
change o(13,occ) 0.0
end
If wanting to do this on more than one atom (e.g., O13 and O9), the command would have been.
\edit
change o(13,occ) 0.0 o(9,occ) 0.0
end
Via the Graphical Interface, put the mouse over the offending atom and "right mouse click"
and select the Edit menu and set the Occupancy to zero. If you wish to fix the parameters for this atom,
put the mouse over the "Unselected" atom and "right mouse click" and go into the
"Sticky refine mode".

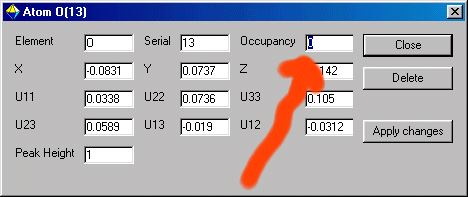
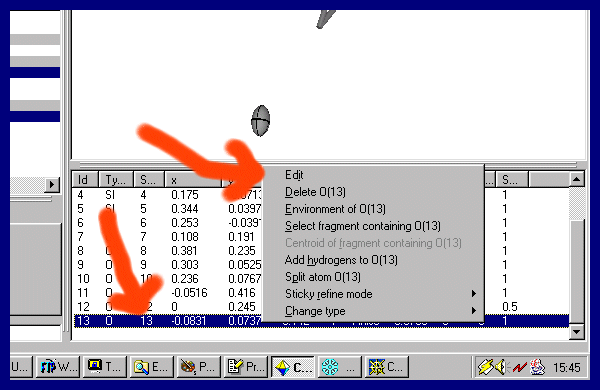
Or you can go to the bottom right Atom List, put the mouse over the offending atom label and "right mouse click"
and select the Edit menu and set the Occupancy to zero. Then Apply the Changes.
If you wish to fix the parameters for this atom,
put the mouse over the "Unselected" atom and "right mouse click" and go into the
"Sticky refine mode".