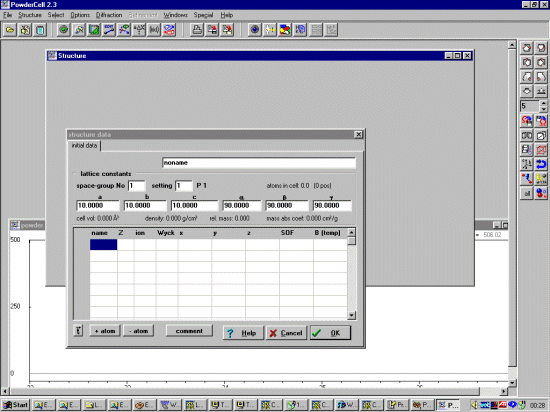
If you enter a new structure from scratch with Powder Cell by selecting File, New, input is done via a menu interface.
Otherwise, Shelx and ICSD files can be used by selecting File, Load.
|
The following run-through is for Powder Cell 2.3
If you enter a new structure from scratch with Powder Cell by selecting File, New, input is done via a menu interface.
Otherwise, Shelx and ICSD files can be used by selecting File, Load.
|
|
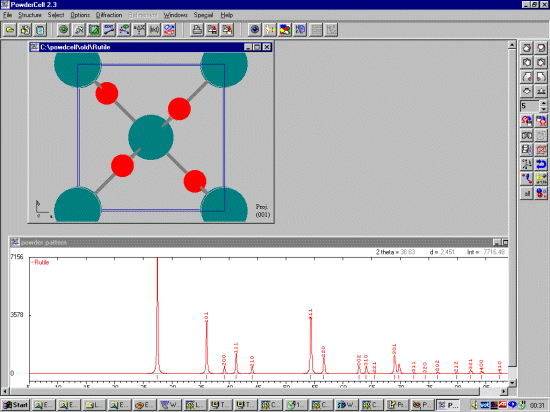
Once a file is selected or a new crystal structure is inputted, Powder Cell
will draw the structure and depending on the saved defaults, calculate a
Powder pattern. Multiple crystal structures can be opened, which can then
be used to explore quantitative analysis and multiphase mixtures.
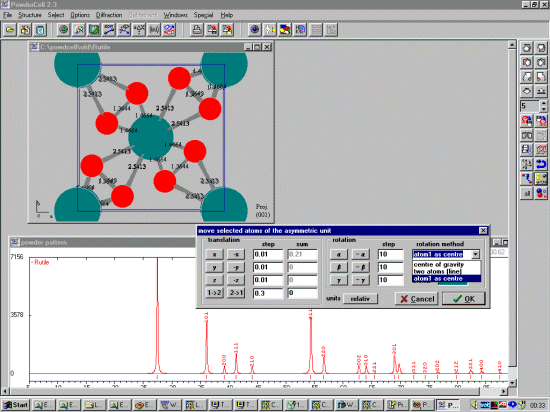
Individual or groups of atoms can be selected via the Select menu, and moved around via the menu interface (Select, Move Selected Atoms); with new powder patterns being calculated on request. The user has the option to display Bond-lengths on the screen at all times.
Phase transitions can be explored using the Structure, Subgroups, Structure, Super Groups menu options. This takes into account all the symmetry requirements of performing such an operation. |
|
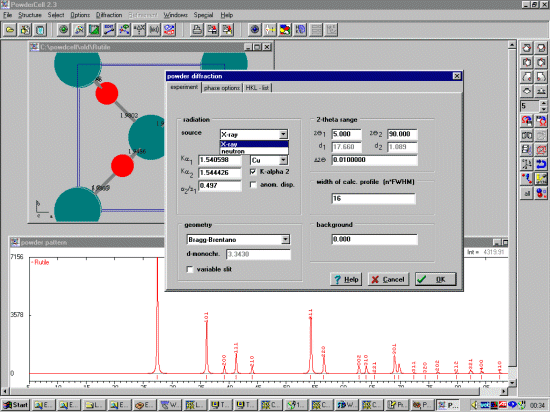
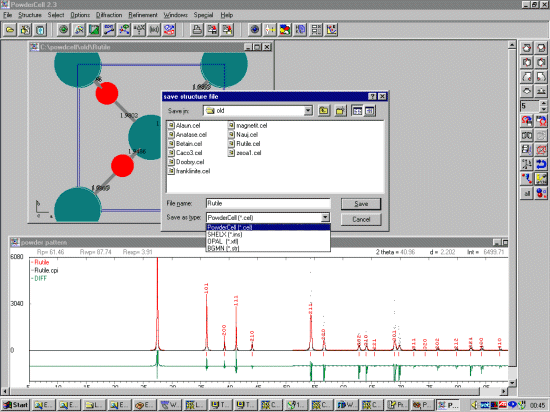
Neutron and X-ray patterns can be calculated; then saved in a variety of
formats including CPI and Siemens/Burker format using the Diffraction,
Export Data menu.
|
|
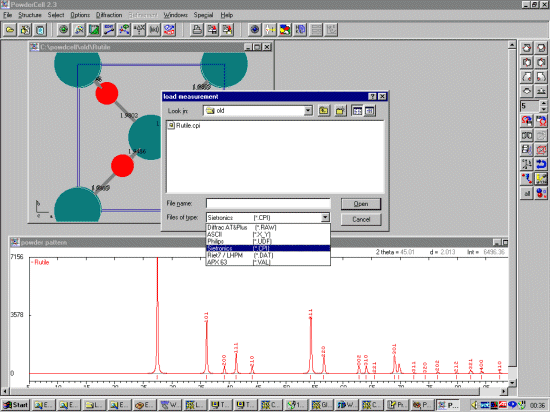
To aid in trialing structures and exploring phase transitions and structure
modifications; it is possible to import "raw diffraction data" in a variety of
data formats.
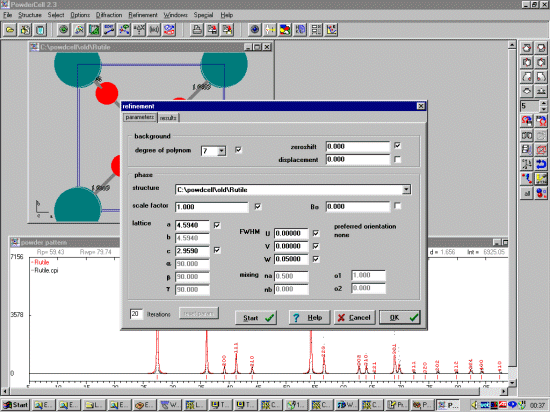
The unit cell and width/shape of the opened crystal structures can then be fitted to the raw diffraction data by using the Refinement menu options.
|
|
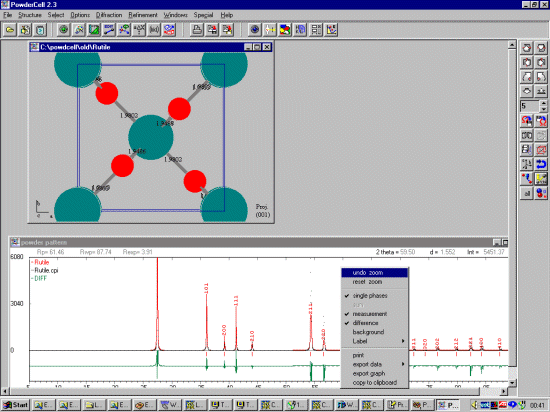
It is possible to enhance the display such as providing a plot of the difference in the
fit. While you have the powder pattern window selected, click on the right mouse
button to see a list of configurable options.
Once you have fitted/refined the cell and width/shape; you can continue trying model structures/changing your structure and seeing how it matches the raw diffraction data. This can be used to examine how structural changes may affect the pattern. Or Powder Cell can be used in this mode to try and obtain a decent starting structure to refine via a Rietveld refinement program.
For continuation via other structure solution and refinement software, Powder Cell can save the structure into a variety of formats include, Shelx, Opal and BGMN Rietveld | [CCP14 BGMN Mirror].
|