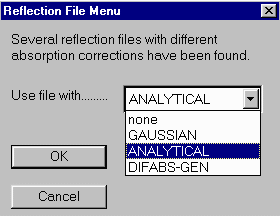
Click on Absorb, RefDelF, DIFABS, General menu option to enter the Difabs absorption correction option.
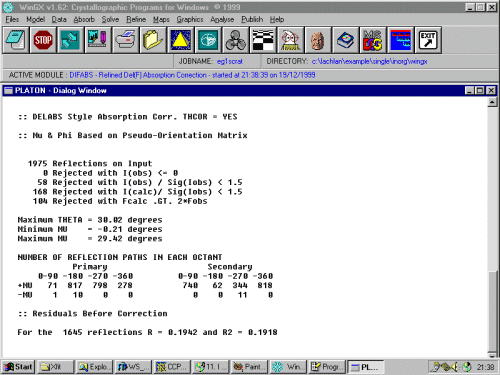
WinGX will pass this over to Platon, where it will generate a output window (as shown below). WinGX may also quickly bring up then quit a Platon absorption surface plot.
After removing the Platon summary screen, WinGX will leave a confirmation dialog box stating .
The thorough Platon output is in the absorb.lst file where you can check the exact corrections Platon has performed on your HKL data. This can be accessed easily via the top menu under Analyse, Open List File, ABSORP
1975 Reflections corrected (Corrections on F**2)
Min. absorption corr. = 0.378 for -16 0 -4 at Phi(P)= 176.4 Mu(P)= 4.2 Phi(S)= 298.9 Mu(S)= 4.2
Max. absorption corr. = 2.103 for 0 0 -2 at PHI(P)= 89.8 MU(P)= 0.0 PHI(S)= 264.1 MU(S)= 0.0
Average absorption correction = 1.017
Minimum, Maximum Virtual Transmission: 0.101 0.564
***VALUES FOR COEFFICIENTS***
0.6365 -0.0138 0.0010 0.2830 0.0040 0.0010
-0.2128 -0.0685 0.2126 0.0774 -0.0081 -0.0134
0.4841 0.1398 -0.0280 -0.1501
-0.0016 -0.0183 -0.0097 0.1744
-0.1732 -0.0187 -0.0024 -0.3789 0.0258 -0.0014 0.0599
0.4957 0.0355 -0.2192 0.0344 -0.0450 0.0860 -0.0889
-0.7614 -0.3010 -0.1200 -0.0308 -0.0602
0.1350 0.2162 0.3040 0.1385 -0.6506
Absorption Surface for PHI = 0 ---> 170 WITH MU RANGE = 0 ---> 30
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
30 1.49 1.63 1.75 1.83 1.85 1.84 1.87 1.94 2.01 2.03 1.95 1.78 1.60 1.43 1.30 1.17 1.03 0.90
25 1.53 1.68 1.80 1.87 1.87 1.85 1.88 1.96 2.06 2.10 2.02 1.85 1.63 1.43 1.26 1.11 0.97 0.84
20 1.58 1.74 1.87 1.93 1.91 1.89 1.91 2.00 2.12 2.17 2.11 1.92 1.68 1.45 1.25 1.07 0.92 0.79
15 1.65 1.82 1.95 2.00 1.98 1.95 1.97 2.06 2.20 2.27 2.22 2.02 1.76 1.49 1.26 1.05 0.88 0.74
10 1.73 1.92 2.06 2.11 2.09 2.05 2.07 2.17 2.31 2.40 2.35 2.15 1.86 1.56 1.29 1.06 0.86 0.71
5 1.84 2.03 2.18 2.25 2.24 2.21 2.24 2.35 2.49 2.58 2.51 2.30 2.00 1.67 1.37 1.10 0.87 0.69
0 2.05 2.20 2.32 2.41 2.48 2.56 2.69 2.86 2.99 2.99 2.82 2.51 2.17 1.85 1.55 1.26 0.98 0.72