This brings up the following screen
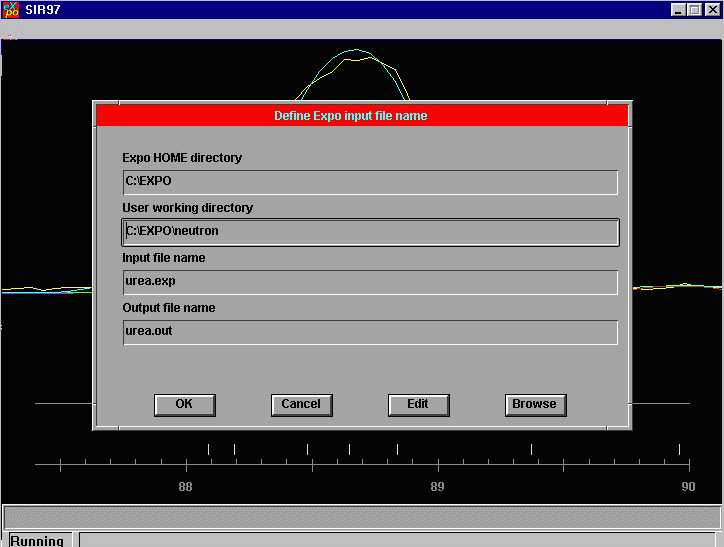
Click on OK and you are then prompted to open up a EXPO control file. If relevant, type in the correct working directory. (in this case c:\expo\neutron)
This example uses calculated Neutron data from a Fullprof Rietveld example file (urea.dat) which consists of deuterated urea.
Also refer to Armel LeBail's Tutorial on solving structures from powder data
Starting InformationThe starting information that is normally known before starting EXPO is:
|
|
From either an Icon in MS-Windows (or typing expo on UNIX
command line), run expo.
This brings up the following screen
Click on OK and you are then prompted to open up a EXPO control file. If relevant, type in the correct working directory. (in this case c:\expo\neutron)
|
|
The file we are going to look at here is described above, urea. Following is
the information inside the file that you would be expect to edit manually and is
described in the EXPO manual
[Home Site] |
[CCP14 Mirror].
One thing to note is the Deuterium scattering factor is defined for H.
%struct urea %job Urea from Fullprof example %init %data neutron format (f9.0) isotope H 0.67 range 5.0000 100.0000 0.0200 pattern urea2.pow cont C 2 O 2 N 4 H 8 wave 1.0791 cell 5.5840 5.584 4.689 90 90 90 space P -4 21 M %extraction %continue Note that you may have to reformat your neutron data to make EXPO happy. A column format seems to be the easiest to make it EXPO happy with the minimum of fuss.
492.000
507.000
461.000
525.000
514.000
467.000
531.000
500.000
532.000
|
|
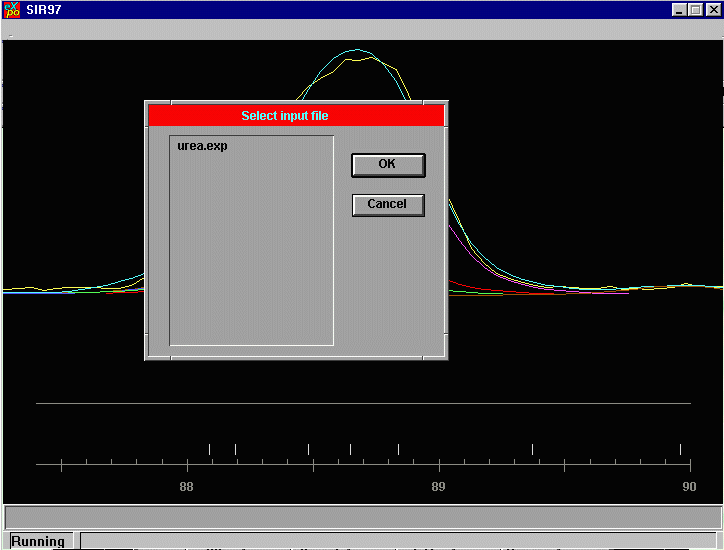
Click on the Browse to view what input files are available
and click on the urea.exp file.
Click on OK to confirm your selection. If the range of the data is incorrect. It could be the format statement in the EXP file is not correct.
|
|
Then click OK again to let EXPO go through its stuff (like Sir97 this is the fun
to watch bit). At various points, the program stops to give you the choice
of changing the defaults. In this example, all you have to do is keep
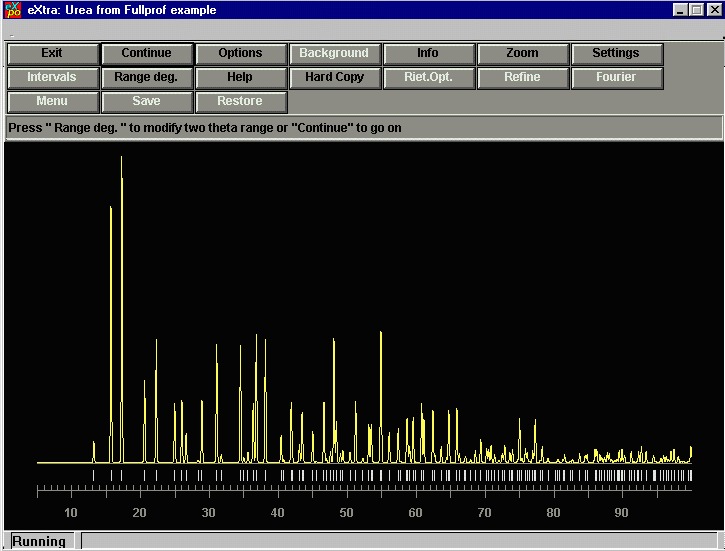
pressing the CONTINUE button. At this point, you can redefine the range of the data on which the structure factor extraction is performed. In this case, we may as well just go up to the full range but in some cases, this is a variable to play with as above a certain angle, the overlap can be so excessive as to be useless.
|
|
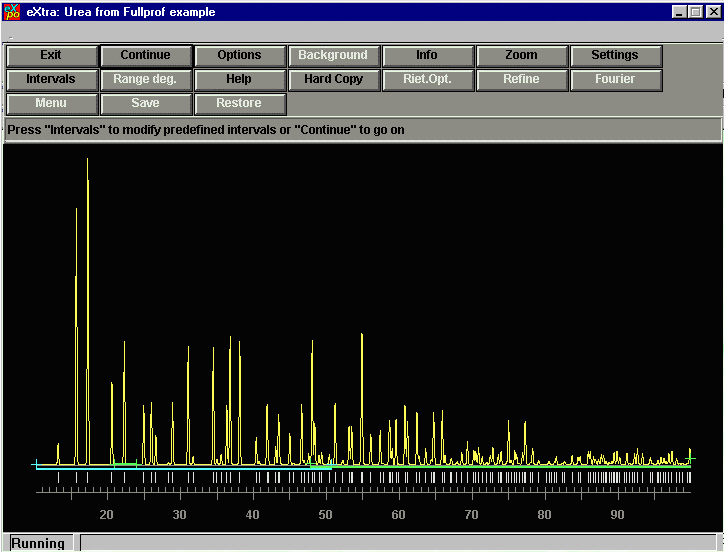
Click continue and EXPO will select what it considers to be the best
intervals for the analysis. You can over-ride this if you which. For this
example, we don't wish to over-ride anything, so just press CONTINUE
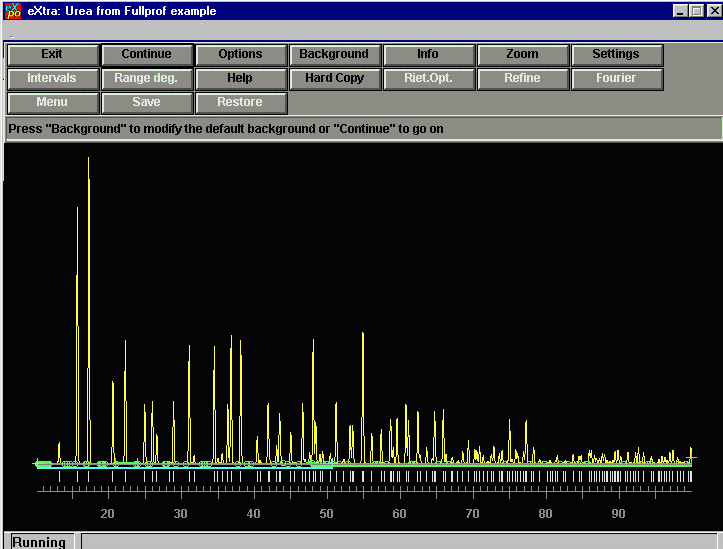
EXPO determines the background. You can zoom and inspect this, but we will just accept this and CONTINUE.
|
|
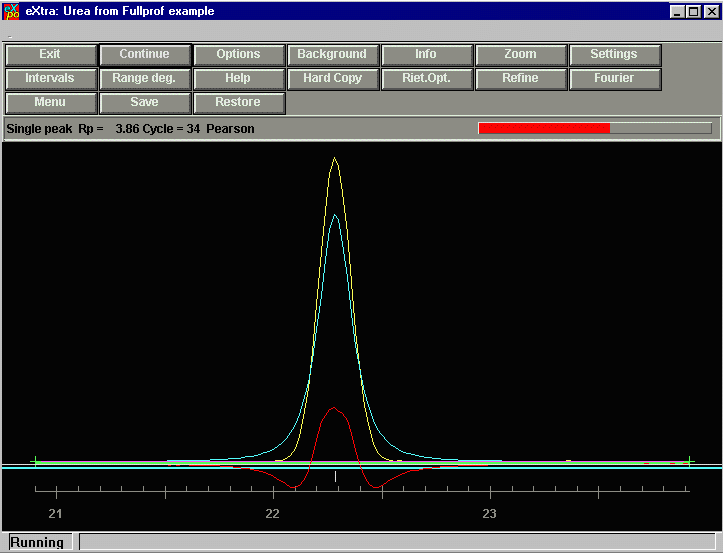
EXPO tries to determine the best peak profile to use, the goes about extracting
intensities in the "regions" it selected.
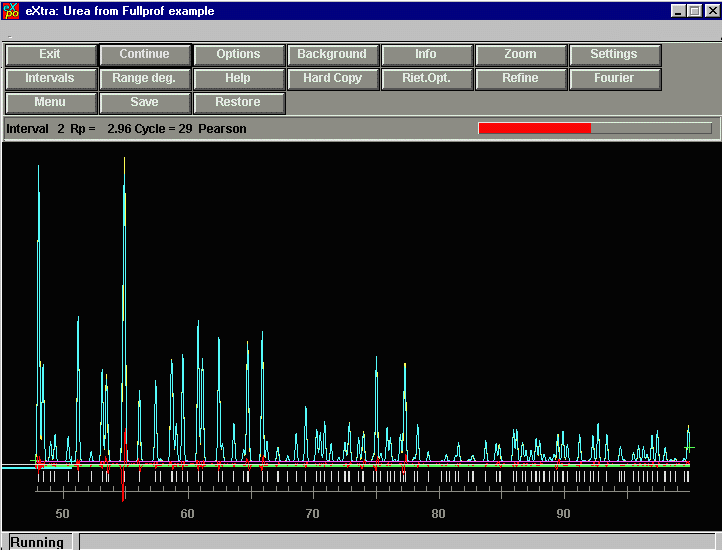
Now it is continuing onto "Interval 2".
Now EXPO does an extraction cycle on the full pattern.
|
|
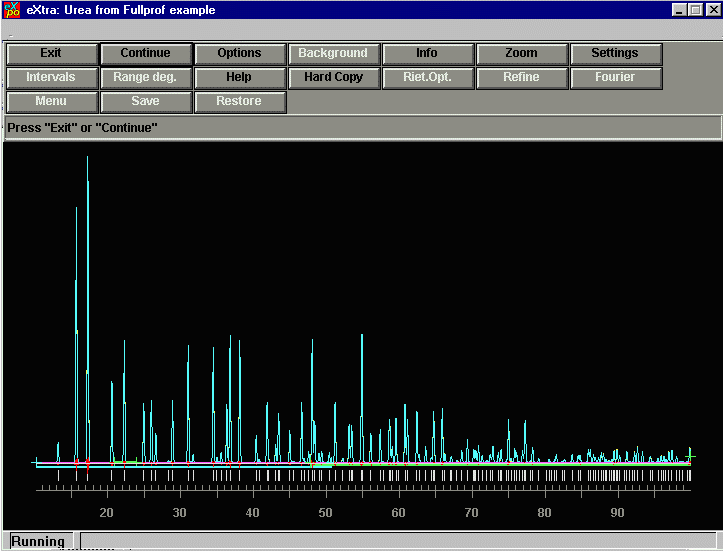
EXPO is now finished the extracting of intensities and now we can
proceed for the direct methods by pressing CONTINUE. In this
case, if EXPO queries if you want to use pseudo-translational effects, just
say no - but normally you would be checking these options out.
|
|
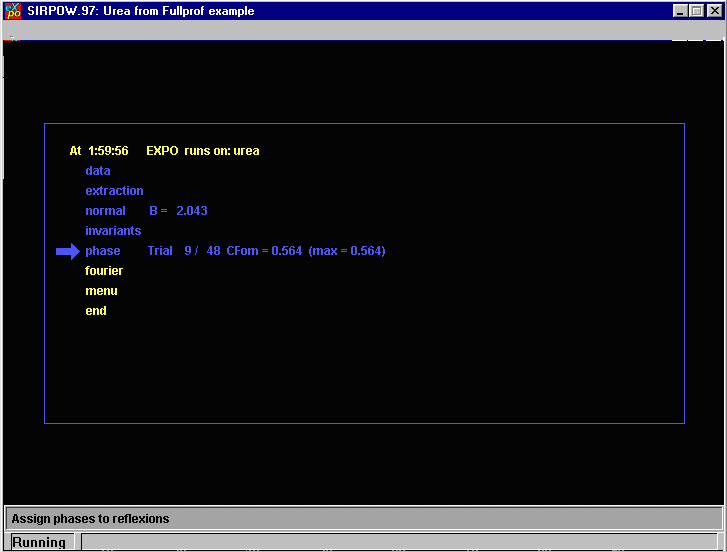
EXPO now is Phasing.
|
|
EXPO is now attempting to place Atoms.
|
|
EXPO now is assigning elements to the atoms.
|
|
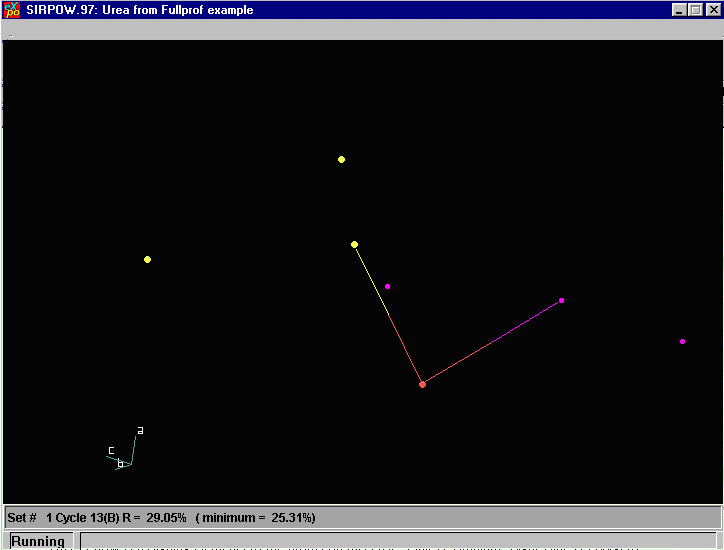
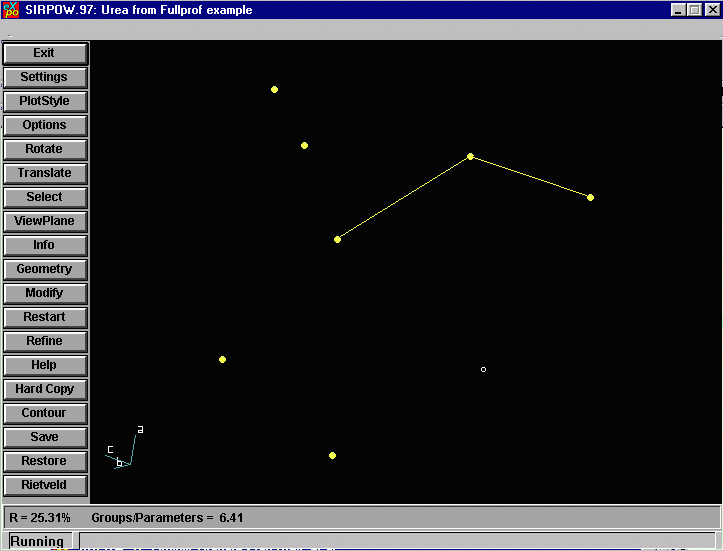
EXPO now presents a structure to us. Note that due to the
nature of neutron diffraction, it may be difficult for both
EXPO and you as the user to define atoms and interpret
the connectivity. Thus, while ESPO may have solved the
structure, the user may have to delve into this and see
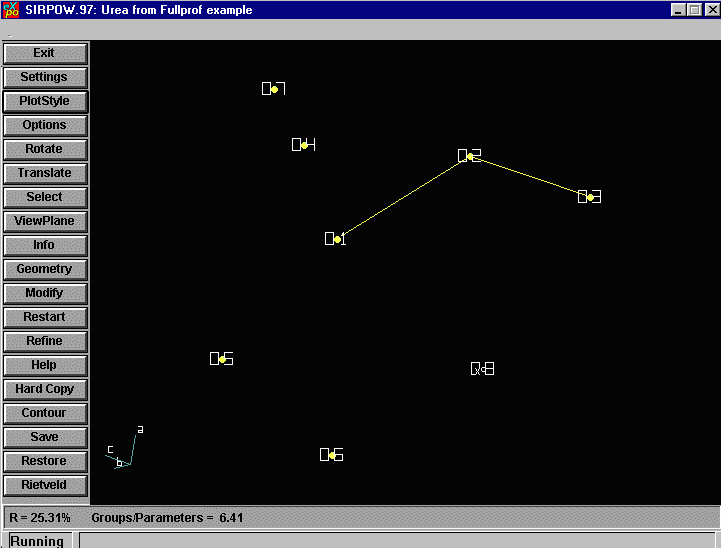
what can be gleaned from the resulting co-ordinates. As with Sir97, we have the option to add Atom labels by using the PlotStyle Menu Option.
|
|
On quitting EXPO produces a *.xyz file with the co-ordinates that can
be input into any other refinement/structure checking program for seeing
if this is a reasonable solution. Bond angles and lengths, as well as
Fourier maps can be examined from within the EXPO menu system.
|
|
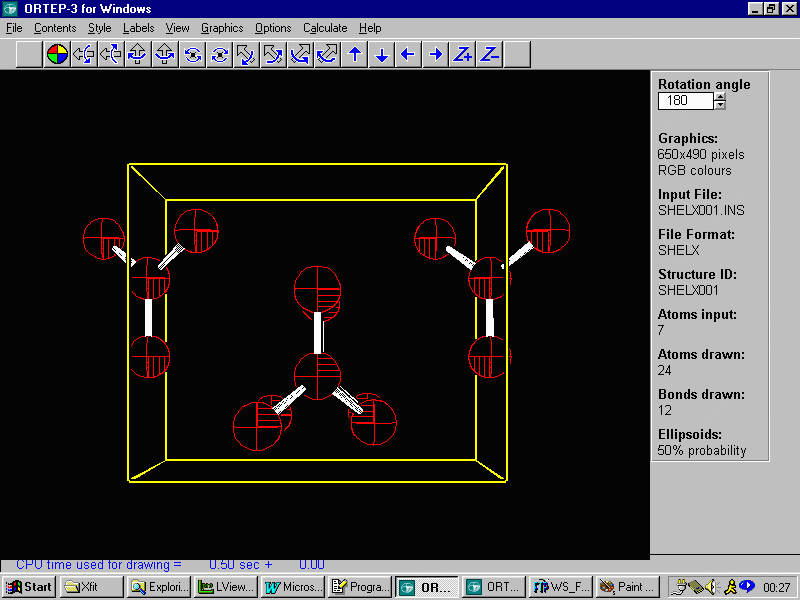
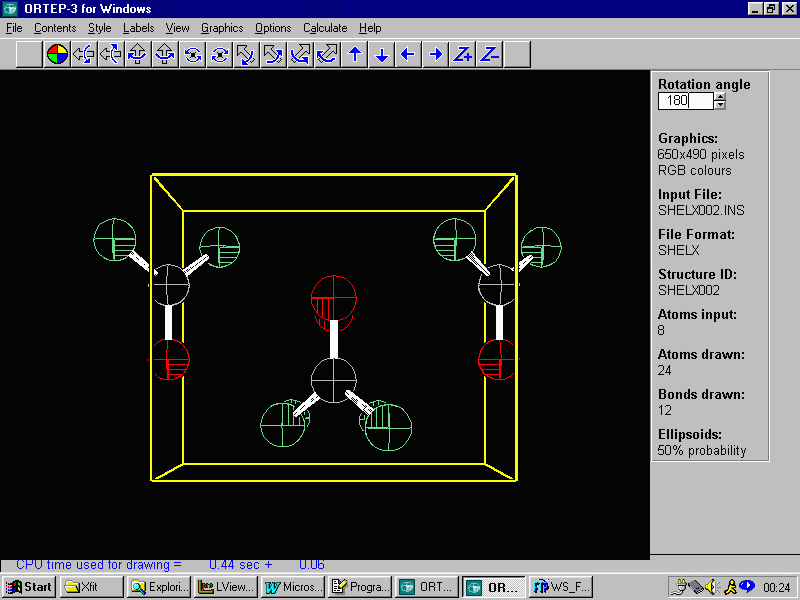
One suggestion it so use Louis Farrugia's WinGX and Ortep-3 to
look and examine the structure. Especially with organics, this may
help resolve connectivity and atom assignment issues. Following
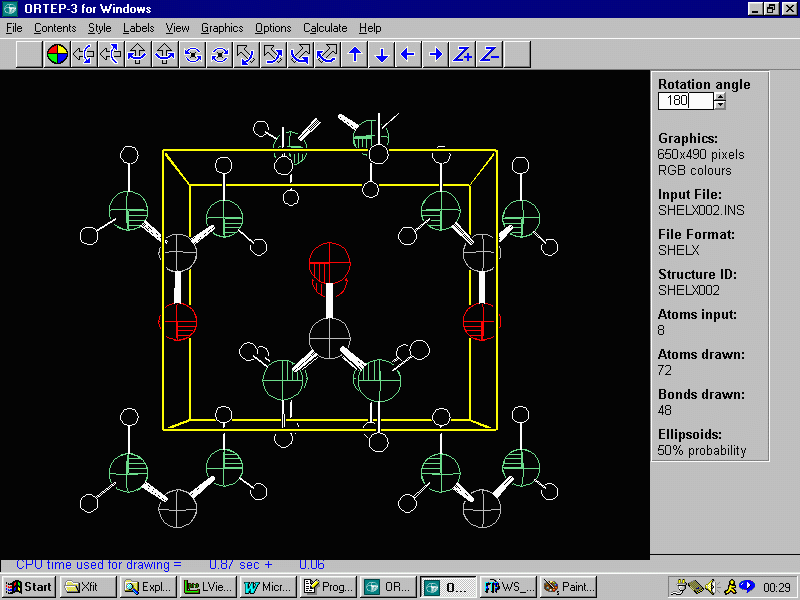
is the EXPO resulting structure less some of the lower peaks. This
is followed by the structure as original defined in Fullprof.
|