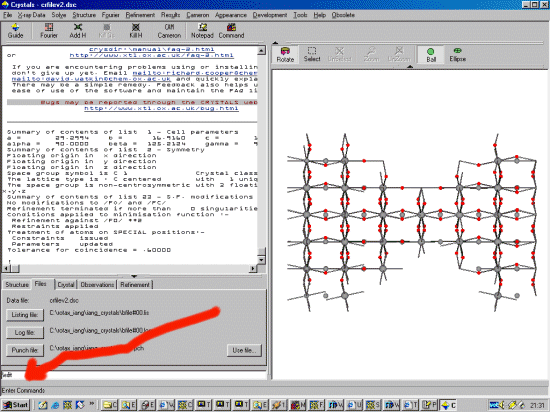
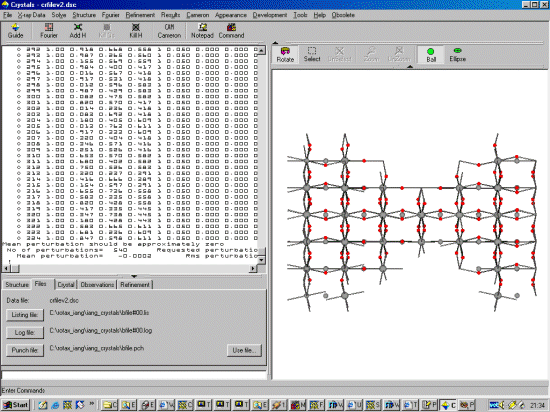
Sometimes, for both refinement or DLS (Distance Least Squares), when a structure has much pseudo symmetry, it can be beneficial to randomly perturb the starting positions of the atoms - especially with hand built "idealised" structure models
Refer:
- Setting up Crystals to perform DLS (Distance Least Squares) on a simple Inorganic Structure
- Setting up Crystals to perform DLS (Distance Least Squares) on an Organic Structure (Cimetidine) to generate an idealised structure from powder diffraction data solved co-ordinates
(Note: Shelx files can be generated from within Crystals using, from the menu, Results, Platon and if you make a mistake, you can use the Multiple UNDO within Crystals to restore from one of the previous structure models.).
The perturbation is in fractions of a unit cell (not in Angstrom), so you just calculate the average perturbation you require based on each differing cell axis.
In this example, an 0.1 Angstrom average perturbation is desired.
Load in the structure and relevant files into crystals (Crystals can import Shelx files). The red arrow points to the crystals "command line" input option which we will be using. (or the following could be made into a script file)