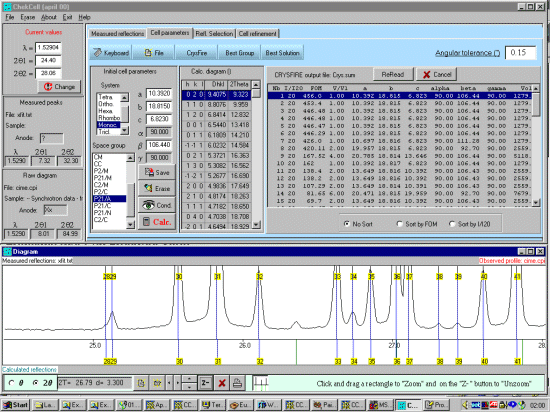
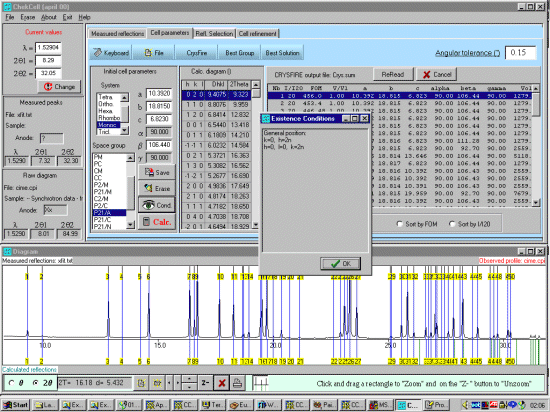
As possibly repeated elsewhere, for organics, it can be best to scope the range of possible results by running ALL the indexing programs (ito, treor, treor, taup, kohl, lzon and fjzn). In this example, this might seem quite contrived as the result is quite obvious (we already know the true answer), but in most cases, being to lax on scoping the possible range of solutions could put you in a world of pain!
For example, on standard laboratory instruments where alignment and sample preparation are of high quality, you might still be lucky to get Figure Of Merits greater than 10 on large cell organics.
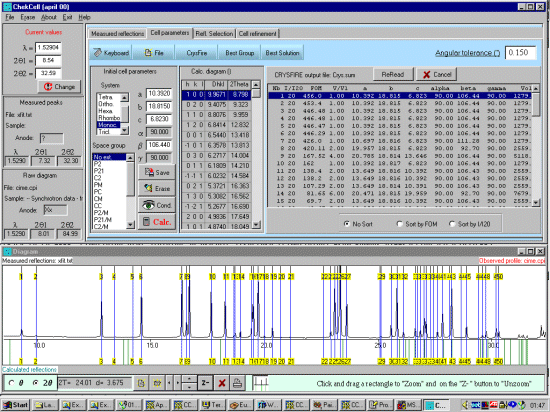
As per the Crysfire tutorials, run XF2CRYS to convert the XFIT.TXT file into a Crysfire *.CDT file. (The wavelength in this case is 1.52904 Angstrom)
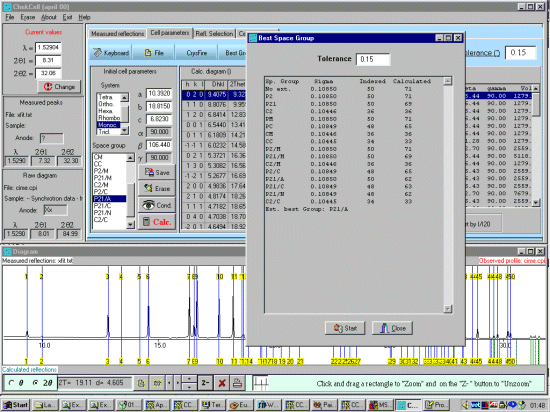
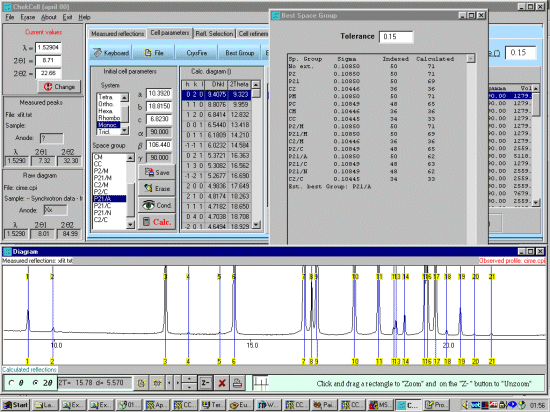
For brevity of this tutorial (though you can be running Chekcell concurrently with Crysfire), run All 7 elligible programs (ito, treor, treor, taup, kohl, lzon and fjzn). Whem prompted, a rule of thumb is to use around 25 peaks, but everyone has their favourite values when these is an excess of peaks to choose from)