Federal Institute for Materials Research and Testing |
|
PowderCell 2.3 — powder pattern calculation from single crystal data and refinement of experimental curves

General
The aim of PowderCell is at first an intuitive structure modification by manipulation step by step and an quasi-simultaneous comparison between experimental data and calculated powder diffraction pattern. At second the program contains refinement procedures to adapt experimental and theoretical data automatically. From our point of view it is recommended to have an user-friendly shell, which is focused on the only necessary input data or the solution of powder diffraction problems exclusively. However, this is not a disadvantage particularly a lot of interesting information the user is able to extract, e.g. concerning crystal symmetry or phase composition, grain size, strain etc.
|
|
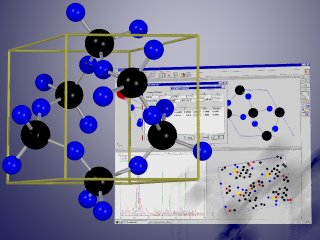
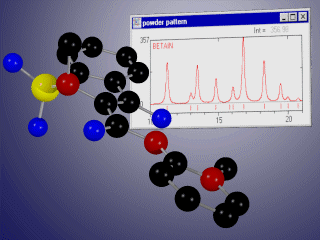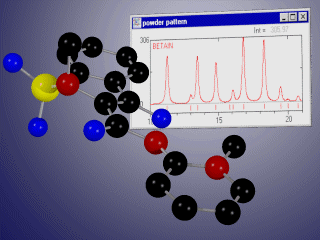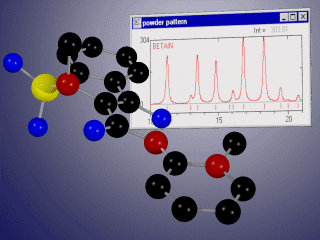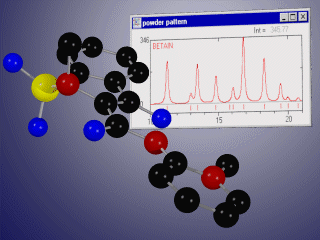
Basic properties
With the help of PowderCell you are e.g. able to
- use different import formats for structure data (ICSD, SHELX, POWDER CELL),
- display crystal structures using more than 740 different settings of space-group types,
- transform the different settings for monoclinic, orthorhombic and rhombohedral space-group types into another,
- generate all klassengleiche and translationengleiche subgroups — an excellent tool for the investigation of phase transitions or other effects described by an decrease of
symmetry density
, - vary the structure arrangement within the unit cell using rotation and translation of atoms or molecules selected before,
- display the corresponding X-ray or neutron powder diffraction patterns simultaneously for up to 10 phases,
- simulate different diffraction conditions, e.g. radiation, doublet splitting, diffraction geometry, variable slits, preferred orientation, anomaleous dispersion, any volume or mass fractions in a phase mixture etc.
- select different convolution functions,
- compare experimental and calculated diffractograms graphically and/or using R-values,
- export the crystal structure and the simulated powder pattern in different graphic formats (e.g. Windows Metafile, PostScript, POVRay),
- use the clipboard to paste graphics and reflection tables into other Windows programs,
- export the diffraction patterns in different file formats (e.g. Siemens Diffrac AT: *.raw).
PowderCell 2.3 offers interesting new features, e.g.
- extended overview of all generated atomic positions, bonding angles and distances, space-group information (general and special positions, Wyckoff notation, list of all maximal subgroups incl. transformation of unit cell axis and origin shift)
- automatical refinement of experimental and simulated powder diffraction data using background polynom, scaling factors, variation of lattice constants, zero shift, reflection broadening, convolution function, preferred orientation...
- calculation of size and strain
- excluded regions in experimental diffraction patterns
- quantitative phase analysis
- decomposition of reflections for an estimation of structure amplitude regarding structure analysis from powder data (LeBail algorithm)



 Back to BAM-I.30 Back to BAM-V.10 Homepage
Back to BAM-I.30 Back to BAM-V.10 Homepage