|
|
What is FFS?
FFS is a GUI (graphic user interface) for the H. Stokes' "Findsym" program, working under Windows platform. It is capable of generating Crystallographic Information Format (*.cif) files from the Findsym output, which can be then easily input into e.g. Rietveld program (GSAS/EXPGUI, Fullprof/EditPcr...). The "automatic assignment" feature works for Atoms/Cryscon input (see below) provided that the user set atom types in the initial Atoms/Cryscon file equal to corresponding atomic numbers.
FFS is also capable of some SIMPLE transformations facilitating the use of Findsym (adding translation, normalization to the supercell etc.).
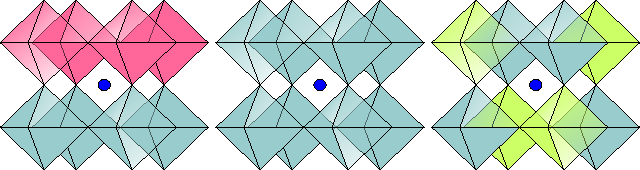
The current version is 1.09R2 @ 03/26/2004. In this version the option to generate (anti)ferroelectric shifts is also incorporated in the GUI mode. This could be useful for modeling of orbital ordering as well.
Possible input formats are:
- Atoms *.atd
("atoms - unit cell, crystal" output ONLY selected) Warning: this option was tested with Atoms 5.0 only!
- "modified Cryscon output" (*.mco) as:
first three lines are comments; all the rest is the list of atoms as in "Cryscon" *.out file ("symmetry-unique
atoms in new unit cell" section).
- (under testing now) Diamond *.csv - generated as
"Display - list - created atoms" / "Save list as - text with separators"
option. In this case the atom types are assigned according to atom
symbolic name in order of their occurrence in the file. Accordingly, the
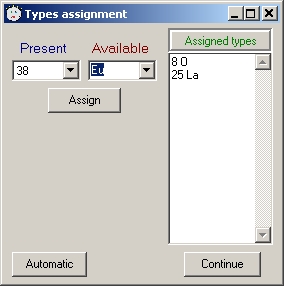
user should manually assign atom types via a GUI when creating *.cif output.
Last Changes
06/29/2004 - version 1.09R3:
- Fullprof-style output of atom list incorporated
(could be useful for those who encountered runtime errors while
trying to import CIF in EditPcr)
03/26/2004 - version 1.09R2:
- Hermann-Mauguin symbol of space group is written in CIF output
03/04/2004 - version 1.09 (beta):
- option to generate (anti)ferroelectric shifts incorporated
01/30/2004 - version 1.08:
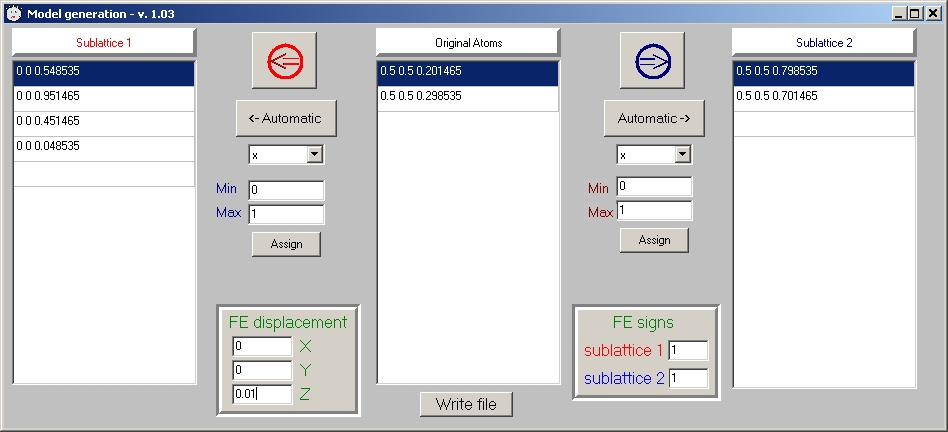
- GUI "model generation" update: allows multiple transferring of atoms with a given value of coordinate or with a coordinate within a specified range
01/28/2004 - version 1.07a:
- CIF output incorporated
- reading lattice parameters from a *.ini file incorporated
User's Guide
In order for the program to work you should have Findsym installed and its directory (c:\isobyu is the default) included in the PATH environment variable.
Below is some explanation on how to use the program. In the case of two sublattices it is possible to use it in a " complete GUI" mode. In the case of more than two sublattices the certain steps involve USER MANUALLY EDITING the input file (*.fsm) using his favorite text editor, e.g. Emacs or Notepad (see p.6 in the example below).
Let us consider an example: we have a structure with one atom of a "sort of interest" (e.g. Mn if you want to model charge ordering in a CMR manganite) in the unit cell, plus some other atoms, that remain disordered in the ordered (CO) structure. We would like to model ordering, that requires 2x2x1 supercell. The sequence of operations is then as follows:
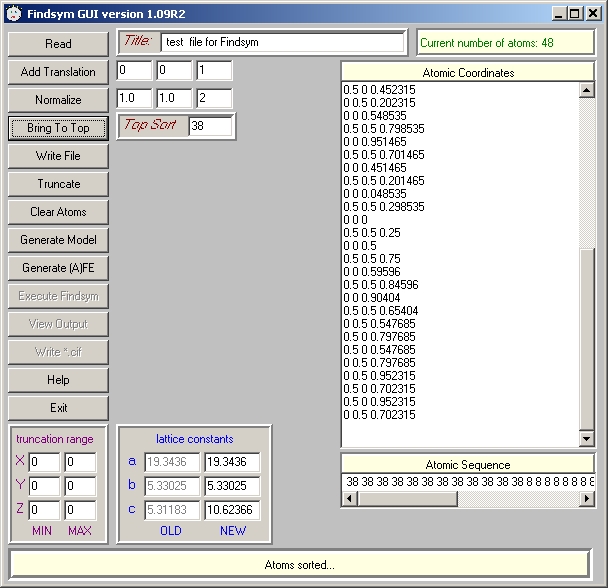
- generate input (by Atoms or Cryscon) - then "Read"
- use "Add translation - 100", "Add translation - 010", "Add translation - 110" - this will fill the required supercell.
- normalize to 221 (Findsym cannot work once the atomic coordinates exceed 1)
- (optional, but convenient) "Bring to top". As a "top sort" specify the type of Mn (or whatever is the atom of interest) as it is in the Atoms/Cryscon file.
- "Write file"
- Now THINK what exactly we are going to model. In the *.fsm file there is a list of atoms with all Mn now being on top (if you did step 4). You should decide what atoms remain equivalent (same charge in case of CO)
and what become different. Then you assign (by modifying line 9 of *.fsm file or using GUI "Generate model" feature in a two-sublattices case) different numbers to those atoms that become inequivalent (e.g. Mn+3 and Mn+4).
- "Execute findsym"
- "View output", "Write *.cif"
It is possible to read lattice parameters from the *.ini file (useful if you need to generate many models from the same input file). If you would like this, put ffs.ini with your values of lattice parameters in the directory where your input file (*.atd, *.mco, *.csv) is located.
Screenshots
Download
Download (free of charge)
FFS (zip, 249 Kb)
Contact
If you find any bugs, have suggestions or would like to incorporate some
other input format (with ALL atoms in the unit cell already in) please
send suggestion, example file and some brief description to Maxim Lobanov (maxl@poltavets.com). However, since it is not my primary research project, I apologize that some of the suggestions could be possibly not introduced or will be somewhat delayed.
|
|
|