Hugo M. Rietveld
As a Ph.D. student at the University of Western Australia between 1961 and 1964, I became thoroughly acquainted with X-ray and neutron diffraction techniques through experiments being conducted at the university and at the HIFAR nuclear reactor in Lucas Heights (NSW). The emphasis was on single crystal diffraction since, even then, the powder method was regarded to be inferior, particularly for structure refinement. During that period the computer entered the scientific field and long, tedious structure factor and density calculations could be obtained more or less instantly. First with the "Mercury" computer at the University of Oxford and the "Siliac" computer at the University of Sydney, and later the IBM 1620 at the Physics Department of the University of Western Australia, computers became an integral part of my crystallography work. They were, incidentally, also important for my later work in the field of powder diffraction.
After obtaining my Ph.D. degree in 1964 (Rietveld 1963), I joined the
neutron diffraction group of the Reactor Centrum Nederland (now Netherlands
Energy Research Foundation ECN) in The Netherlands. This group had only just
been established and was principally engaged in the construction of a neutron
powder diffractometer. The emphasis here was mainly on powder diffraction
techniques, because it was apparent that no large, single crystals could be
grown of the materials that were then of interest. The first crystal
structures to be determined were rather simple and of a high symmetry, with
the result that the peaks were well resolved and integrated intensities could
easily be obtained for further refinement. However, with compounds more
complex and of lower symmetry, the overlap of peaks became so severe that
separating them became practically impossible. In an effort to overcome this
problem, the resolution of the diffractometer was significantly increased by
using a wavelength of ![]() and by eliminating the higher order
wavelength
with a filter of pyrolytic graphite (Loopstra 1966). This proved to be of
appreciable value, especially for structure determination. For structure
refinement, however, the increase of resolution certainly resulted in a
better defined pattern, but often not to such an extent that the peaks were
completely resolved. The solution then was to refine by using overlapping
intensities (Rietveld 1966a). This worked well, but the fact remained that
all extra information contained in the profile of these overlapping peaks was
lost. The following step was to separate the overlapping peaks by trying to
fit Gaussian peaks using least squares procedures. This method also had its
limitations, however, and did not work for severe overlap.
and by eliminating the higher order
wavelength
with a filter of pyrolytic graphite (Loopstra 1966). This proved to be of
appreciable value, especially for structure determination. For structure
refinement, however, the increase of resolution certainly resulted in a
better defined pattern, but often not to such an extent that the peaks were
completely resolved. The solution then was to refine by using overlapping
intensities (Rietveld 1966a). This worked well, but the fact remained that
all extra information contained in the profile of these overlapping peaks was
lost. The following step was to separate the overlapping peaks by trying to
fit Gaussian peaks using least squares procedures. This method also had its
limitations, however, and did not work for severe overlap.
Before the advent of computers, data reduction was a must in crystallography
in order to be able to handle a relatively complex structure. Integrated
intensities were therefore the smallest data elements one could work with
practically. To consider using the individual intensities, ![]() ,
constituting a
step-scanned diffraction peak as data was completely unrealistic. With the
experience of using computers for single crystal structure refinements and
having seen their enormous capacity for handling large amounts of data, I saw
that the spectre of increasing the number of data by a factor of ten by using
the individual intensities,
,
constituting a
step-scanned diffraction peak as data was completely unrealistic. With the
experience of using computers for single crystal structure refinements and
having seen their enormous capacity for handling large amounts of data, I saw
that the spectre of increasing the number of data by a factor of ten by using
the individual intensities, ![]() , instead of the integrated intensities,
constituted no real barrier. In the first refinement program, the intensities
, instead of the integrated intensities,
constituted no real barrier. In the first refinement program, the intensities
![]() were corrected for background and were read in together with the value
of
the relative contributions each constituent peak made, i.e. the value of
were corrected for background and were read in together with the value
of
the relative contributions each constituent peak made, i.e. the value of
![]() in the expression
in the expression
![]() (Rietveld 1967), where S is the structure factor.
(Rietveld 1967), where S is the structure factor.
These values were calculated from the unit-cell dimensions and the
wavelength, and zeropoint and halfwidth values measured directly from the
diagram. Also, for resolved peaks the integrated values were used rather than
the ![]() intensities, because the Gaussian peak shape did not fit well at
lower
angles. Later, a correction for the asymmetry was introduced. The non-
refinement of the reflection-profile parameters can be explained by the fact
that the then available computer, the Electrologica X1, was not powerful
enough to solve a least squares problem of more than a very limited number of
parameters. With the arrival of the larger Electrologica X8 computer, the
program was rewritten to include the capability of refining structure as well
as profile parameters (Rietveld 1969b). Twenty-seven copies of this program,
written in Algol (Rietveld 1969b) and later in 1972, in FORTRAN IV, were
distributed to institutes all over the world and this has greatly contributed
to the acceptance of the method.
intensities, because the Gaussian peak shape did not fit well at
lower
angles. Later, a correction for the asymmetry was introduced. The non-
refinement of the reflection-profile parameters can be explained by the fact
that the then available computer, the Electrologica X1, was not powerful
enough to solve a least squares problem of more than a very limited number of
parameters. With the arrival of the larger Electrologica X8 computer, the
program was rewritten to include the capability of refining structure as well
as profile parameters (Rietveld 1969b). Twenty-seven copies of this program,
written in Algol (Rietveld 1969b) and later in 1972, in FORTRAN IV, were
distributed to institutes all over the world and this has greatly contributed
to the acceptance of the method.
The method was first reported at the seventh Congress of the IUCr in Moscow in 1966 (Rietveld 1966b). The response was slight, or, rather, non-existent, and it was not until the full implementation of the method was published (Rietveld 1969b), that reactions came. At this time, the method was mainly used to refine structures from data obtained by fixed wavelength neutron diffraction; a total of 172 structures were refined in this way before 1977 (Cheetham and Taylor 1977). In the previously mentioned paper (Rietveld 1969b), it had been suggested that the method could also be applied to X-ray data, but it was not until 1977 (Malmros and Thomas 1977; Young et al. 1977; Khattak and Cox 1977) that the method became generally accepted for X-ray as well as neutron powder diffraction, first with fixed wavelength and then also with fixed angle (energy dispersive data). This is reflected in an increasing number of citations to the original papers (Rietveld 1967 and 1969b) as published in the Science Citation Index. Figure 1 shows the number of citations between the years 1967 and 1994.
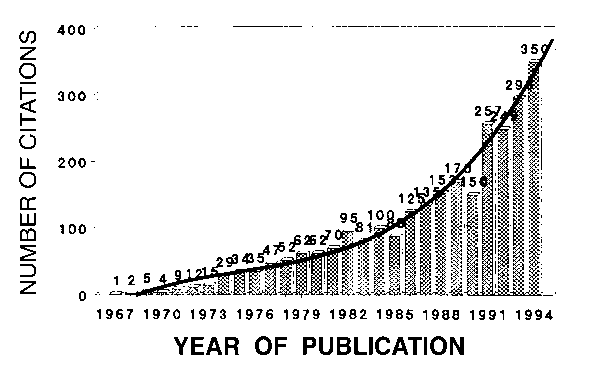
Fig. 1. Number of publications, in the Science Citation Index, citing as reference the original papers (Rietveld 1967, 1969b) on the Rietveld method or having the name Rietveld in their title.
In the period January 1987 to May 1989 a total of 341 papers were published
with reference to or using the Rietveld Method, of which nearly half used
neutron diffraction. The review article of Albinati and Willis (1982) gives
a good impression of the state of the method at that moment. Many more papers
on the method have appeared since, often with unexpected applications.
It has been most gratifying for me to experience how the Rietveld Method has contributed to a renewed interest in powder diffraction techniques, even to the extent that in some applications it replaces single crystal techniques. The method is proven to be sound and has given results at least as good as single crystal data. The possible under-estimation of the actual probable errors by the calculated estimated standard deviations, may serve as a reminder to all users that the method is not to be treated as a black box. One must be continually aware of the limitations, not only of this method, but in general of all least squares methods. In this respect I fully agree with Prince (1981) who states that "If the fit of the assumed model is not adequate, the precision and accuracy of the parameters cannot be validly assessed by statistical methods".