|
|
Don't worry, Endeavour has been specifically designed to keep this point as simple as possible.
Even without any knowledge of structural modeling you will be able to provide good parameters for most
crystal structures !
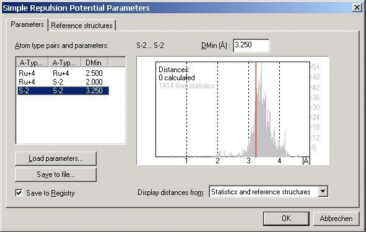
This is especially true for the default simple repulsion potential: Its parameters are simply minimum
interatomic distances for all atom type pairs in your compound. For example, if your compound is RuS2,
you will have to provide minimum distance values between Ru-Ru, Ru-S and S-S.
|
|
|
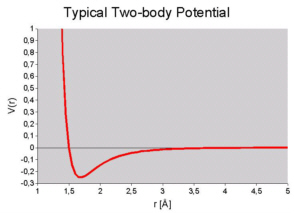
It seems that with the Endeavour concept (simultaneous optimization of pattern difference and potential energy)
a new problem arises: How to calculate the potential energy ?
Ab-initio calculations are still far too slow to be used by Endeavour (though this would definitely be desirable...).
Hence, Endeavour has to use empirical two-body potential functions. However, these functions need certain
parameters which unfortunately have to be provided by the user (at least at the current state of research...).
Don't start to look for your chemistry textbook now; Endeavour displays distance statistics for each atom type
pair over the complete ICSD database (ICSD = Inorganic Crystal Structure Database). All you have to do is adjust
the slider to best match the distance distribution displayed in the background for this specific atom type pair.
That's it !
And for most conventional organic molecular structures it is even more simple:
Reasonable parameters for the recommended simple repulsion potential are already present
in Endeavour's internal database, so the user does'nt have to worry about parameter settings
at all in these cases !
|