|
Step 6 of 7:
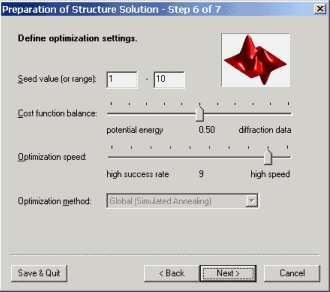
Define optimization settings
This page offers some options concerning the optimization. For instance, the
so-called 'cost function balance' may be adjusted, i.e. the contribution of
either of the two parts of the cost function to the overall value. Thus, weak
diffraction data may to a certain degree be balanced by a high quality
potential and vice versa.
Besides this, the speed of the calculation is controlled by the slider
'optimization speed' that could take any integer value from 1 to 10. A value of
10 means highest optimization speed, however, because of the underlying
simulated annealing optimization method, also lowest success probability.
|